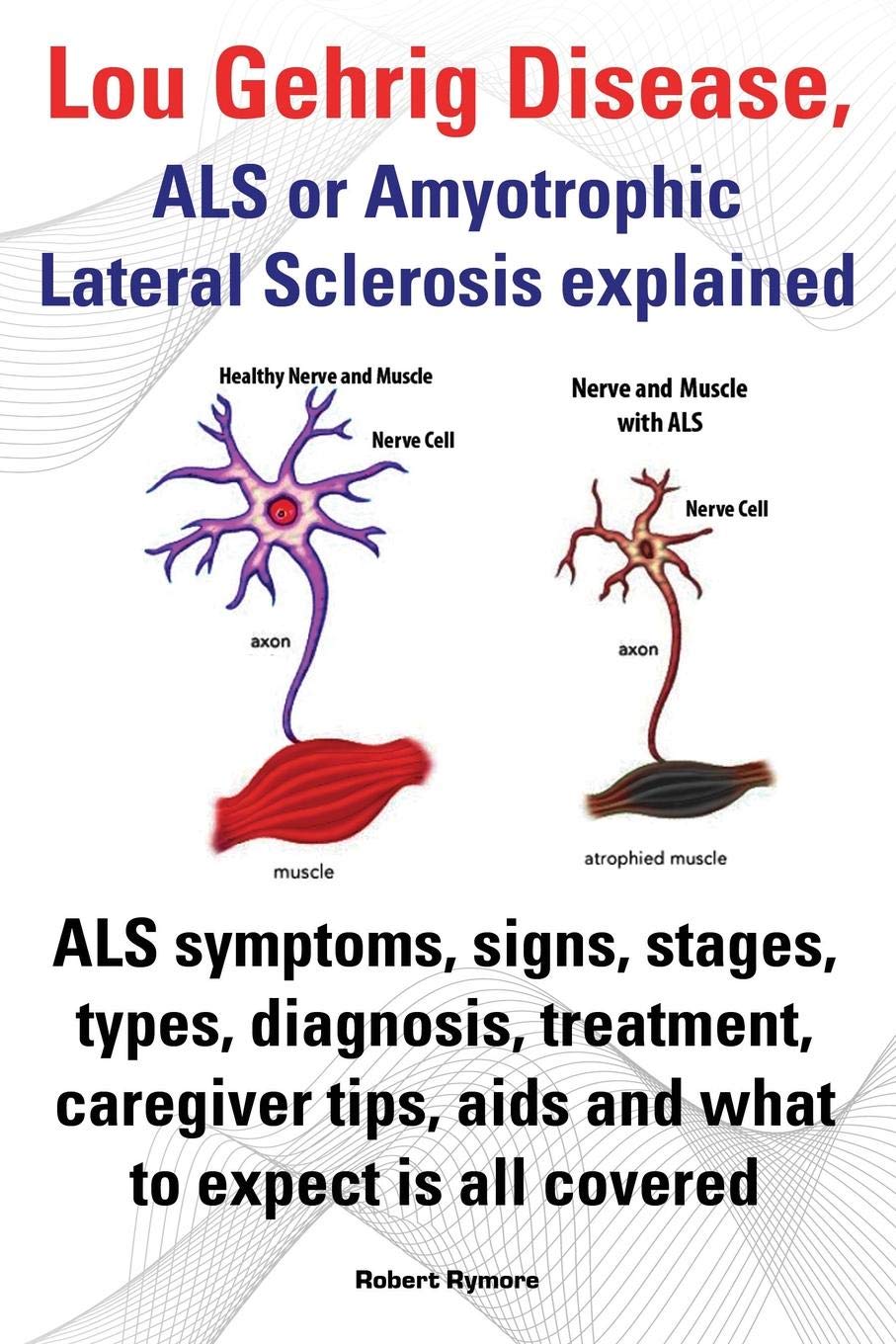
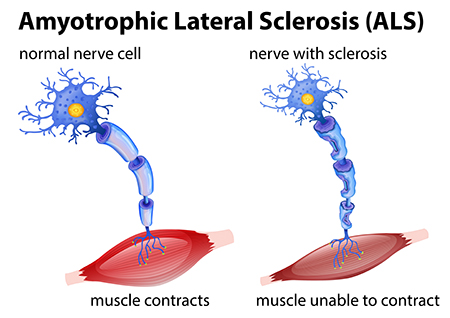
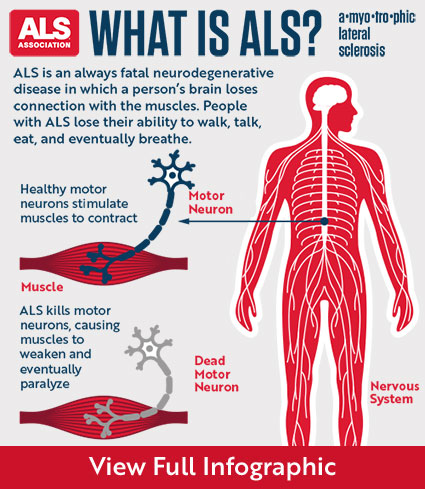
What disorders are related to ASD?
Certain known genetic disorders are associated with an increased risk for autism, including Fragile X syndrome (which causes intellectual disability) and tuberous sclerosis (which causes benign tumors to grow in the brain and other vital organs) — each of which results from a mutation in a single, but different, gene. Recently, researchers have discovered other genetic mutations in children diagnosed with autism, including some that have not yet been designated as named syndromes. While each of these disorders is rare, in aggregate, they may account for 20 percent or more of all autism cases.
People with ASD also have a higher than average risk of having epilepsy. Children whose language skills regress early in life — before age 3 — appear to have a risk of developing epilepsy or seizure-like brain activity. About 20 to 30 percent of children with ASD develop epilepsy by the time they reach adulthood. Additionally, people with both ASD and intellectual disability have the greatest risk of developing seizure disorder.
How is ASD diagnosed?
ASD symptoms can vary greatly from person to person depending on the severity of the disorder. Symptoms may even go unrecognized for young children who have mild ASD or less debilitating handicaps. Very early indicators that require evaluation by an expert include:
- no babbling or pointing by age 1
- no single words by age 16 months or two-word phrases by age 2.
- no response to name
- loss of language or social skills previously acquired
- poor eye contact
- excessive lining up of toys or objects
- no smiling or social responsiveness
Later indicators include:
- impaired ability to make friends with peers
- impaired ability to initiate or sustain a conversation with others
- absence or impairment of imaginative and social play
- repetitive or unusual use of language
- abnormally intense or focused interest
- preoccupation with certain objects or subjects
- inflexible adherence to specific routines or rituals
Health care providers will often use a questionnaire or other screening instrument to gather information about a child’s development and behavior. Some screening instruments rely solely on parent observations, while others rely on a combination of parent and doctor observations. If screening instruments indicate the possibility of ASD, a more comprehensive evaluation is usually indicated.
A comprehensive evaluation requires a multidisciplinary team, including a psychologist, neurologist, psychiatrist, speech therapist, and other professionals who diagnose and treat children with ASD. The team members will conduct a thorough neurological assessment and in-depth cognitive and language testing. Because hearing problems can cause behaviors that could be mistaken for ASD, children with delayed speech development should also have their hearing tested.
What causes ASD?
Scientists believe that both genetics and environment likely play a role in ASD. There is great concern that rates of autism have been increasing in recent decades without full explanation as to why. Researchers have identified a number of genes associated with the disorder. Imaging studies of people with ASD have found differences in the development of several regions of the brain. Studies suggest that ASD could be a result of disruptions in normal brain growth very early in development. These disruptions may be the result of defects in genes that control brain development and regulate how brain cells communicate with each other. Autism is more common in children born prematurely. Environmental factors may also play a role in gene function and development, but no specific environmental causes have yet been identified. The theory that parental practices are responsible for ASD has long been disproved. Multiple studies have shown that vaccination to prevent childhood infectious diseases does not increase the risk of autism in the population.
What role do genes play?
Twin and family studies strongly suggest that some people have a genetic predisposition to autism. Identical twin studies show that if one twin is affected, then the other will be affected between 36 to 95 percent of the time. There are a number of studies in progress to determine the specific genetic factors associated with the development of ASD. In families with one child with ASD, the risk of having a second child with the disorder also increases. Many of the genes found to be associated with autism are involved in the function of the chemical connections between brain neurons (synapses). Researchers are looking for clues about which genes contribute to increased susceptibility. In some cases, parents and other relatives of a child with ASD show mild impairments in social communication skills or engage in repetitive behaviors. Evidence also suggests that emotional disorders such as bipolar disorder and schizophrenia occur more frequently than average in the families of people with ASD.
In addition to genetic variations that are inherited and are present in nearly all of a person’s cells, recent research has also shown that de novo, or spontaneous, gene mutations can influence the risk of developing autism spectrum disorder. De novo mutations are changes in sequences of deoxyribonucleic acid or DNA, the hereditary material in humans, which can occur spontaneously in a parent’s sperm or egg cell or during fertilization. The mutation then occurs in each cell as the fertilized egg divides. These mutations may affect single genes or they may be changes called copy number variations, in which stretches of DNA containing multiple genes are deleted or duplicated. Recent studies have shown that people with ASD tend to have more copy number de novo gene mutations than those without the disorder, suggesting that for some the risk of developing ASD is not the result of mutations in individual genes but rather spontaneous coding mutations across many genes. De novo mutations may explain genetic disorders in which an affected child has the mutation in each cell but the parents do not and there is no family pattern to the disorder. Autism risk also increases in children born to older parents. There is still much research to be done to determine the potential role of environmental factors on spontaneous mutations and how that influences ASD risk.
Do symptoms of autism change over time?
For many children, symptoms improve with age and behavioral treatment. During adolescence, some children with ASD may become depressed or experience behavioral problems, and their treatment may need some modification as they transition to adulthood. People with ASD usually continue to need services and supports as they get older, but depending on severity of the disorder, people with ASD may be able to work successfully and live independently or within a supportive environment.
How is autism treated?
There is no cure for ASD. Therapies and behavioral interventions are designed to remedy specific symptoms and can substantially improve those symptoms. The ideal treatment plan coordinates therapies and interventions that meet the specific needs of the individual. Most health care professionals agree that the earlier the intervention, the better.
Educational/behavioral interventions: Early behavioral/educational interventions have been very successful in many children with ASD. In these interventions therapists use highly structured and intensive skill-oriented training sessions to help children develop social and language skills, such as applied behavioral analysis, which encourages positive behaviors and discourages negative ones. In addition, family counseling for the parents and siblings of children with ASD often helps families cope with the particular challenges of living with a child with ASD.
Medications: While medication can’t cure ASD or even treat its main symptoms, there are some that can help with related symptoms such as anxiety, depression, and obsessive-compulsive disorder. Antipsychotic medications are used to treat severe behavioral problems. Seizures can be treated with one or more anticonvulsant drugs. Medication used to treat people with attention deficit disorder can be used effectively to help decrease impulsivity and hyperactivity in people with ASD. Parents, caregivers, and people with autism should use caution before adopting any unproven treatments