What is ITP?
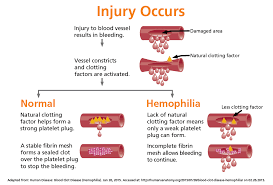
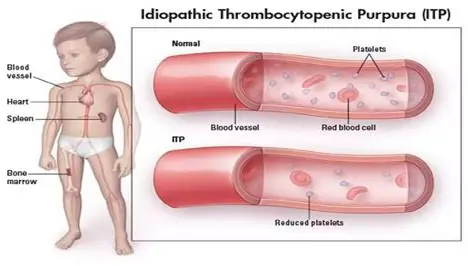
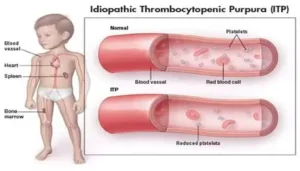
ITP means idiopathic thrombocytopenic purpura which is an autoimmune disease. The immune system is mistakenly attacking and destroying good platelets. In autoimmune diseases, the body mounts an immune attack toward one or more seemingly normal organ systems. In ITP, platelets are the target. They are marked as foreign by the immune system and eliminated in the spleen, the liver, and by other means. In addition to increased platelet destruction, some people with ITP also have impaired platelet production.
A normal platelet count is between 150,000 and 400,000/microliter of blood. If someone has a platelet count lower than 100,000/microliter of blood with no other reason for low platelets, that person is considered to have ITP.1 There is no accurate, definitive test to diagnose ITP.
SYMPTOMS:
Simple to understand. Platelets are for clotting our blood; if the platelet count is high we clot too much if low, in ITP, we bleed easy to hemorrage.
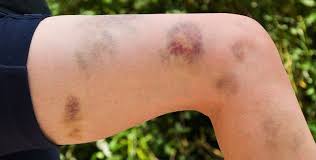
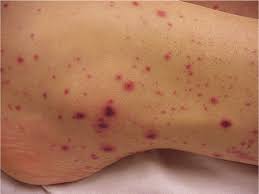
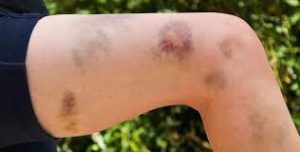
With few platelets, people with ITP often have bleeding symptoms such as spontaneous bruising, petechiae (pe-TEEK-ee-ay), tiny red dots on the skin, Bleeding from the gums or nose, and for women, possibly heavy menses. More severe bleeding symptoms include blood blisters on the inside of the mouth, blood in the urine or stool, or bleeding in the brain.
Idiopathic thrombocytopenic purpura or immune thrombocytopenia affects children and adults. Children often develop ITP after a viral infection and usually recover fully without treatment. In adults, the disorder is often long term.
Treatments for the disease vary depending on the platelet count, severity of symptoms, age, lifestyle, personal preferences, and any other associated diseases. Some people may choose to not treat their disease and live with low platelets.
While it may seem like ITP is a simple disease, there are nuances to the diagnosis, differences in the disease between children and adults, and variations in how the disease responds to treatments.
TYPES OF ITP:
Newly diagnosed ITP: within 3 months from diagnosis
Persistent ITP: 3 to 12 months from diagnosis. During this phase, patients have not reached spontaneous remission or maintained a complete response off therapy
Chronic ITP: lasting for more than 12 months
Severe ITP: presence of bleeding symptoms that need treatment or need an increase from prior treatment
Refractory ITP: does not respond or is resistant to attempted forms of treatment
RISK FACTORS:
-Your sex. Women are two to three times more likely to develop ITP than men are.
-Recent viral infection. Many children with ITP develop the disorder after a viral illness, such as mumps, measles or a respiratory infection.
COMPLICATIONS:
-A rare complication of ITP, bleeding into the brain, which can be fatal.
-Pregnancy
In pregnant women with ITP, the condition doesn’t usually affect the baby. But the baby’s platelet count should be tested soon after birth.
If you’re pregnant and your platelet count is very low or you have bleeding, you have a greater risk of heavy bleeding during delivery.
DIAGNOSIS:
1. M.D. will exclude other possible causes of bleeding and a low platelet count, such as an underlying illness or medications being the cause of low platelet count, not ITP.
2. Take a history of the child or adult, including their family.
3. Complete blood count (CBC). Looks at red blood, white blood and platelet cells counts.
4 Blood smear. This test is often used to confirm the number of platelets observed in a complete blood count.
5.Bone marrow exam. This test may be used to help identify the cause of a low platelet count, though the American Society of Hematology doesn’t recommend this test for children with ITP. All cells (platelets) are produced in the bone marrow. Bone marrow will be normal because a low platelet count is caused by the destruction of platelets in the bloodstream and spleen — not by a problem with the bone marrow.
TREATMENT:
People with mild idiopathic thrombocytopenic purpura may need nothing more than regular monitoring and platelet checks. Children usually improve without treatment. Most ITP adults will eventually need treatment as it gets worse or becomes chronic.
1-The M.D will stop any meds that inhibit platelet production=Anti-platelet Meds (Ex. aspirin, ibuprofen (Advil, Motrin IB, others), ginkgo biloba and warfarin, also known as Coumadin)
2-Drugs that suppress your immune system. M.D. might start you on oral corticosteroid, such as prednisone and when platelet count is normal gradually decrease the dosing till no longer on it. The problem is that many adults experience a relapse after stopping corticosteroids. A new course of corticosteroids may be pursued, but long-term use of these medications is unusual, due to its long term side effects. These include cataracts, high blood sugar, increased risk of infections and thinning of bones (osteoporosis).
3-Injections to increase your blood count (Ex. immune globulin (IVIG). This drug may also be used if you have critical bleeding or need to quickly increase your blood count before surgery. The effect usually wears off in a couple of weeks.
4-Drugs that boost platelet production. Examples romiplostim (Nplate) and eltrombopag (Promacta) — help your bone marrow produce more platelets.
5-Other immune-suppressing drugs. Rituximab (Rituxan) helps reduce the immune system response that’s damaging platelets, thus raising the platelet count.
6-Removal of your spleen.
7-Other drugs. Azathioprine (Imuran, Azasan) has been used to treat ITP. But it can cause significant side effects.
Review all treatments with your personal doctor.