Spina Bifida is the most common permanently disabling birth defect in the United States.
80% of these are located in the lumbar and sacral areas (lower back) of the spine.
Spina Bifida literally means “split spine.”
Spina Bifida happens when a baby is in the womb and the spinal column does not close all of the way. Every day, about 8 babies born in the United States have Spina Bifida or a similar birth defect of the brain and spine.
Spina bifida occurs during the third and fourth weeks of pregnancy when a portion of the fetal spinal cord fails to properly close. As a result, the child is born with a part of the spinal cord exposed on the back.
No one knows for sure the exact cause of spina bifida but have their ideas. Scientists believe that genetic and environmental factors act together to cause the condition.
Spina bifida is a birth defect that mainly affects the spine. Normally in the first month of pregnancy, a special set of cells forms the “neural tube.” The top of the tube becomes the brain and the remainder becomes the spinal cord and structures around it. In spina bifida, the neural tube doesn’t close all the way and some of the bones of the spine don’t close in the back.
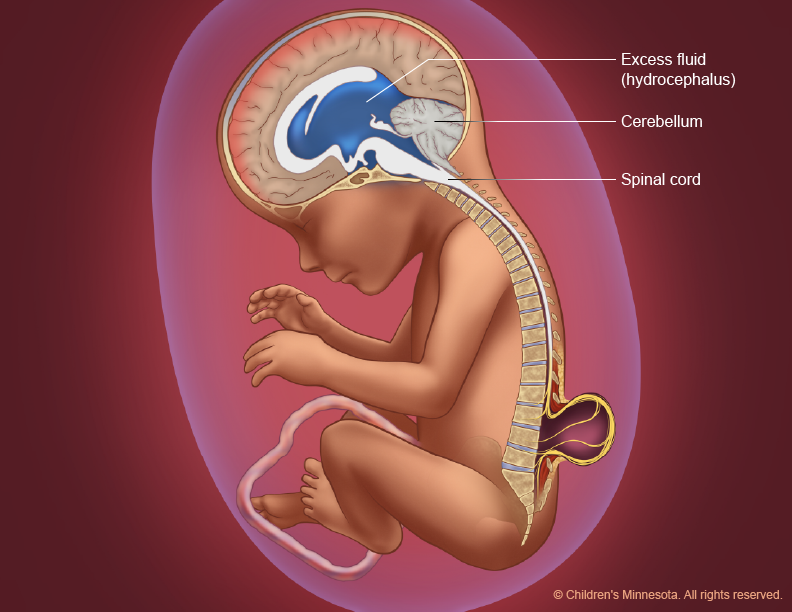
Often, abnormalities of the brain (such as hydrocephalus, described below) accompany abnormalities of the spine because the neural tube closes first in the middle and then closure proceeds both upward and downward—meaning that if something happens that prevents normal formation of the spine, it may also prevent normal formation of the part of the brain that is forming (closing) at the same time.
The term neural tube defect describes a group of conditions, including spina bifida, that occur when the neural tube does not close all the way.
Although doctors and researchers don’t know for sure why spina bifida occurs, they have identified a few risk factors:
- Race. Spina bifida is more common among whites and Hispanics.
- Sex. Girls are affected more often.
- Family history of neural tube defects. Couples who’ve had one child with a neural tube defect have a slightly higher chance of having another baby with the same defect. That risk increases if two previous children have been affected by the condition.
- In addition, a woman who was born with a neural tube defect, or who has a close relative with one, has a greater chance of giving birth to a child with spina bifida. However, most babies with spina bifida are born to parents with no known family history of the condition.
- Folate deficiency. Folate (vitamin B-9) is important to the healthy development of a baby. Folate is the natural form of vitamin B-9. The synthetic form, found in supplements and fortified foods, is called folic acid. A folate deficiency increases the risk of spina bifida and other neural tube defects.
- Some medications. Anti-seizure medications, such as valproic acid (Depakene), seem to cause neural tube defects when taken during pregnancy, perhaps because they interfere with the body’s ability to use folate and folic acid.
- Diabetes. Women with diabetes who don’t control their blood sugar well have a higher risk of having a baby with spina bifida.
- Obesity. Pre-pregnancy obesity is associated with an increased risk of neural tube birth defects, including spina bifida.
- Increased body temperature. Some evidence suggests that increased body temperature (hyperthermia) in the early weeks of pregnancy may increase the risk of spina bifida. Elevating your core body temperature, due to fever or the use of saunas or hot tubs, has been associated with increased risk of spina bifida.
- If you have known risk factors for spina bifida, talk with your doctor to determine if you need a larger dose or prescription dose of folic acid, even before a pregnancy begins.
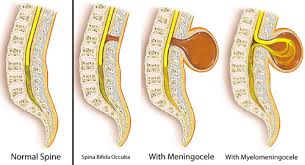
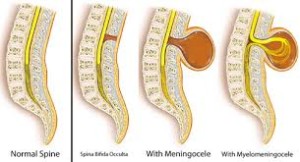
There are 4 different types of Spina Bifida:
occulta, closed neural tube defects, meningocele, and myelomeningocele.
1-Occult Spinal Dysraphism (OSD) Infants with this have a dimple in their lower back. Because most babies with dimples do not have OSD, a doctor has to check using special tools and tests to be sure. Other signs are red marks, hyperpigmented patches on the back, tufts of hair or small lumps. In OSD, the spinal cord may not grow the right way and can cause serious problems as a child grows up. Infants who might have OSD should be seen by a doctor, who will recommend tests.
2-Spina Bifida Occulta It is often called “hidden Spina Bifida” because about 15 % of healthy people have it and do not know it. Spina Bifida Occulta usually does not cause harm, & has no visible signs. Spinal Cord & nerves are usually fine.
Visible indications of spina bifida occulta (SBO) can sometimes be seen on the newborn’s skin above the spinal defect, including:
- An abnormal tuft of hair
- A collection of fat
- A small dimple or birthmark. Meningocele A meningocele causes part of the spinal cord to come through the spine like a sac that is pushed out. Nerve fluid is in the sac, and there is usually no nerve damage. Individuals with this condition may have minor disabilities.
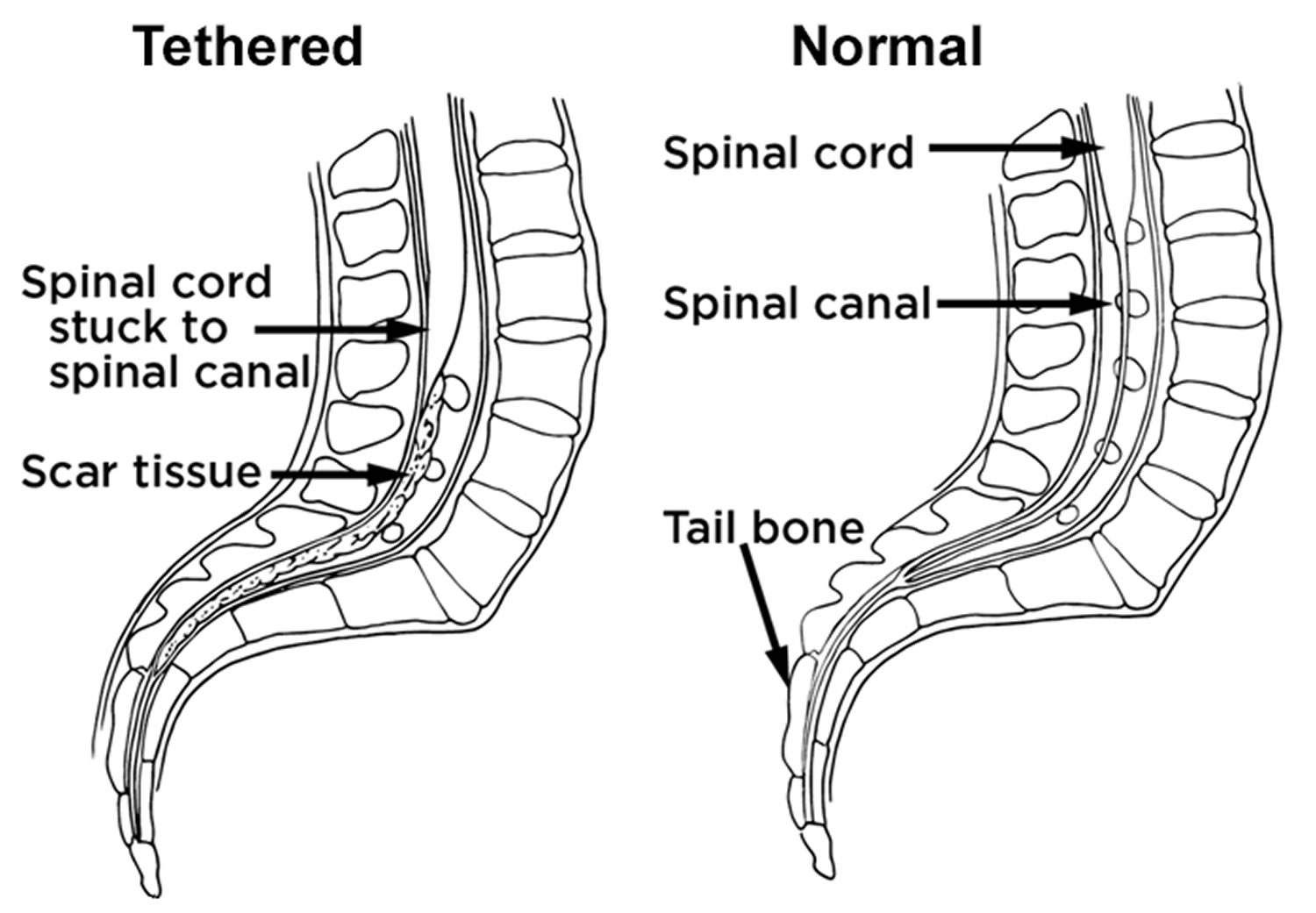
- Many people who have spina bifida occulta don’t even know it, unless the condition is discovered during an X-ray or other imaging test done for unrelated reasons. People find out they have it after having an X-ray of their back. It is considered an incidental finding because the X-Ray is normally done for other reasons. However, in a small group of people with SBO, pain and neurological symptoms may occur. Tethered cord can be an insidious complication that requires investigation by a neurosurgeon.
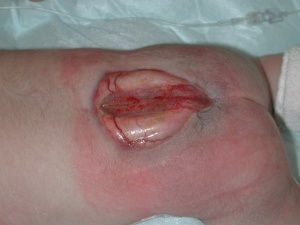
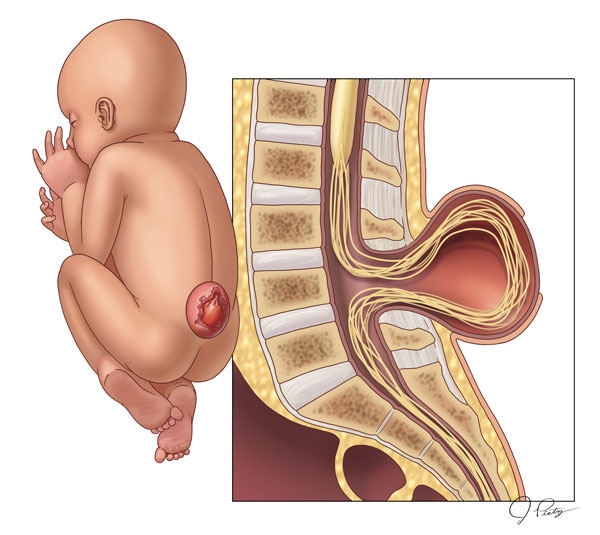
3-Meningomyelocele is a type of spina bifida. Spina bifida is a birth defect in which the spinal canal and the backbone don’t close before the baby is born. This type of birth defect is also called a neural tube defect. Meningocele occurs when the bones do not close around the spinal cord and the meninges are pushed out through the opening, causing a fluid-filled sac to form. The meninges are three layers of membranes covering the spinal cord, consisting of dura mater, arachnoid mater and pia mater. In most cases, the spinal cord and the nerves themselves are normal or not severely affected. The sac is often covered by skin and may require surgery. This is the rarest type of spina bifida.
4-Myelomeningocele (Meningomyelocele), also called Spina Bifida Cystica. Myelomeningocele is the most severe form of spina bifida, occurring nearly once for every 1,000 live births.
Number 4 – This is the most severe form of Spina Bifida.
It happens when parts of the spinal cord and nerves come through the open part of the spine. It causes nerve damage and other disabilities. 70 to 90% of children with this condition also have too much fluid on their brains HYDROCEPHALUS. This happens because fluid that protects the brain and spinal cord is unable to drain like it should. The fluid builds up, causing pressure and swelling. Without treatment, a person’s head grows too big, and may have brain damage. Children who do not have Spina Bifida can also have this problem, so parents need to check with a doctor. Usually, however, tissues and nerves are exposed, making the baby prone to life-threatening infections. A portion of the spinal cord or nerves is exposed in a sac through an opening in the spine that may or may not be covered by the meninges. The opening can be closed by surgeons while the baby is in utero or shortly after the baby is born. Myelomeningocele is often called a snowflake condition because no two people with the condition are the same. Typically, the lower in the spine the opening occurs relates to less symptoms in the person. People with myelomeningocele require close follow-up with physicians throughout their childhood and lifespan to maximize their function and prevent complications such as kidney failure.
Neurological impairment is common, there is brain structure changes including:
- Muscle weakness of the legs, sometimes involving paralysis
- Bowel and bladder problems
- Seizures, especially if the child requires a shunt
- Orthopedic problems — such as deformed feet, uneven hips and a curved spine (scoliosis)Treatment for spina bifida depends on the severity of the condition.
In general, Spina Bifida Treatment:
- Most people with spina bifida occulta require no treatment at all.
- Children with meningocele typically require surgical removal of the cyst and survive with little, if any, disability.
- Children with myelomeningocele, however take the longest road with treatment. It require complex and often lifelong treatment and assistance. Almost all of them survive with appropriate treatment starting soon after birth. Their quality of life depends at least partially on the speed, efficiency, and comprehensiveness with which that treatment is provided. A child born with myelomeningocele requires specialty care. If the hospital the baby is in does not have newborn neuro surgery than the following is done:
- The child should be transferred immediately to a center where newborn surgery can be performed.
- Treatment with antibiotics is started as soon as the myelomeningocele is recognized; this prevents infection of the spinal cord, which can be fatal.
The operation involves closing the opening in the spinal cord and covering the cord with muscles and skin taken from either side of the back.
Remember the most common complications are tethered spinal cord and hydrocephalus, which can have very severe consequences. This is discussed in the next couple of topics!
Updated 10/19/2021
 SPINA BIFIDA
SPINA BIFIDA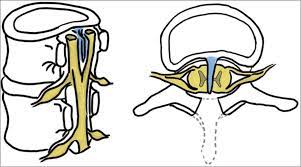
 HYDROCEPHALUS
HYDROCEPHALUS