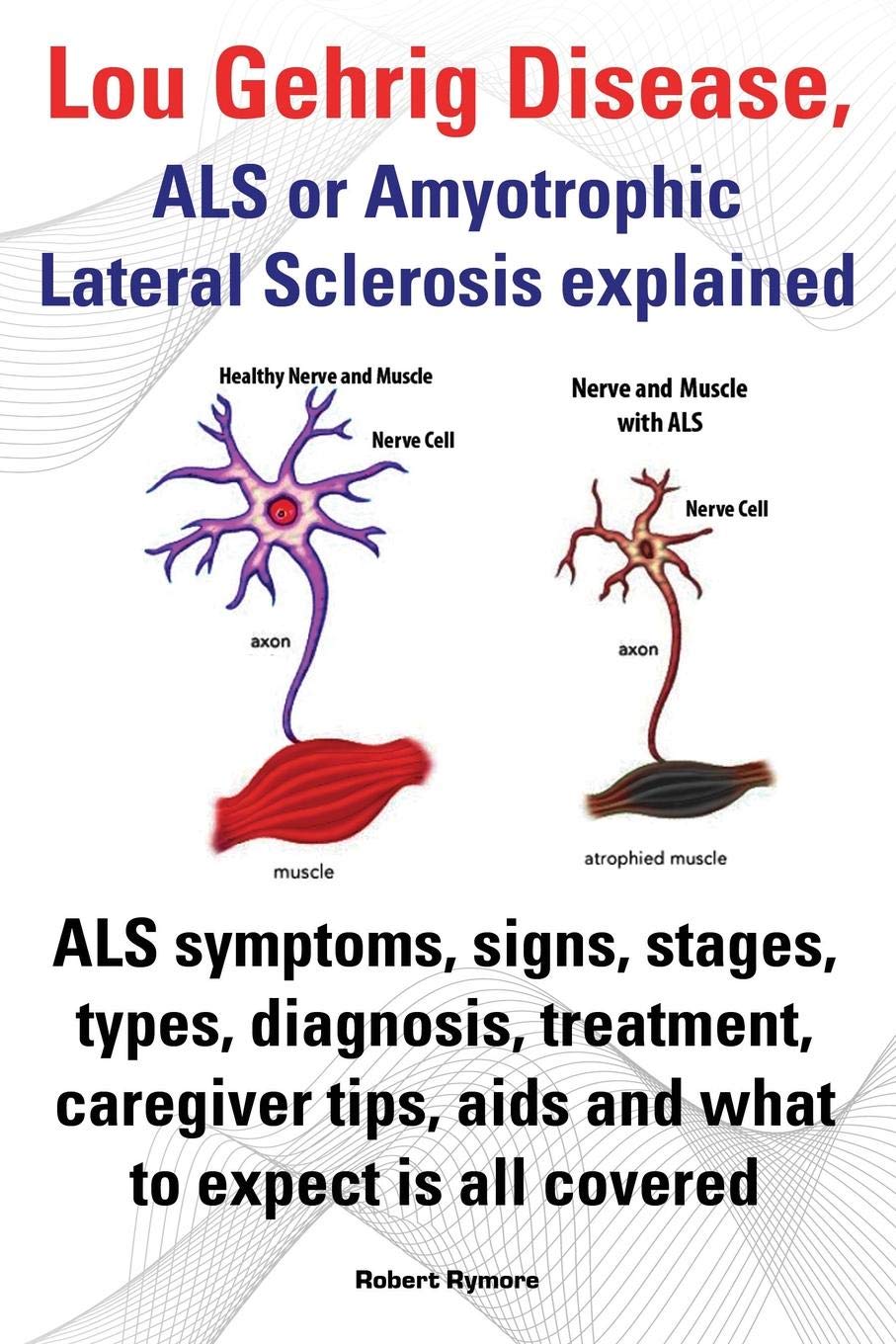

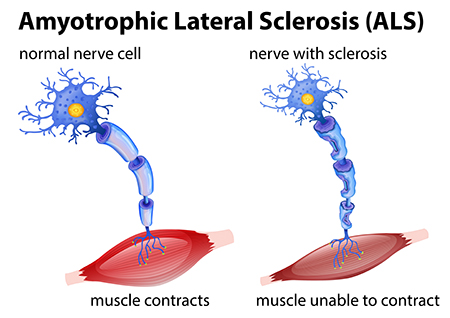
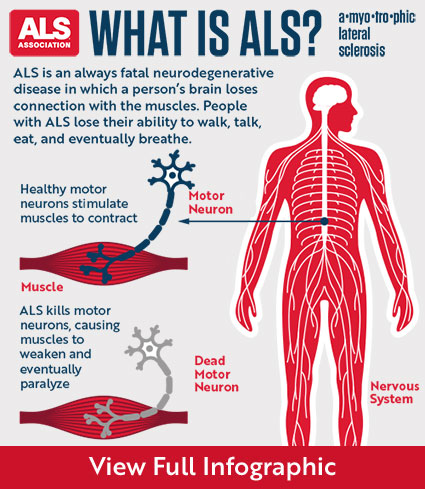
Once ALS starts, it almost always progresses, eventually taking away the ability to walk, dress, write, speak, swallow, and breathe, and shortening the life span. The onset of ALS often involves muscle weakness or stiffness as early symptoms. Progression of weakness, wasting, and paralysis of the muscles of the limbs and trunk, as well as those that control vital functions such as speech, swallowing, and breathing, generally follows.
How fast and in what order this occurs is very different from person to person. While the average survival time is three years, about 20 percent of people with ALS live five years, 10 percent will survive 10 years and 5 percent will live 20 years or longer.
End stages of ALS often include trouble swallowing and speaking. Weak and paralyzed vocal cords make speaking difficult and eventually impossible. Trouble swallowing occurs gradually in some patients, but can occur suddenly in others.
Stages of ALS
ALS is a relentlessly progressive disorder. The rate of progression between individuals is variable and the history generally reflects gradual and progressive worsening over time until death occurs.
Early stages:
Muscles
- Muscles may be weak and soft, or they may be stiff, tight, and spastic. Muscle cramping and twitching (fasciculation) occurs, as does loss of muscle bulk (atrophy).
- Symptoms may be limited to a single body region or mild symptoms may affect more than one region.
Physical effects
- The person may experience fatigue, poor balance, slurred words, a weak grip, tripping when walking, or other minor symptoms.
- Sometimes this stage occurs before a diagnosis is made.
Middle stages:
Muscles
- Symptoms become more widespread.
- Some muscles are paralyzed, while others are weakened or unaffected. Fasciculations may continue.
Physical effects
- Unused muscles may cause contractures, in which the joints become rigid, painful, and sometimes deformed.
- If a fall occurs, the person may not be able to stand back up alone.
- Driving is relinquished.
- Weakness in swallowing muscles may cause choking and greater difficulty eating and managing saliva.
- Weakness in breathing muscles can cause respiratory insufficiency, especially when lying down.
- Some people experience bouts of uncontrolled and inappropriate laughing or crying (pseudobulbar affect). Despite how it seems, the person usually doesn’t feel particularly sad or happy.
Late stages:
Muscles
- Most voluntary muscles are paralyzed.
- The muscles that help move air in and out of the lungs are severely compromised.
Physical effects
- Mobility is extremely limited, and help is needed in caring for most personal needs.
- Poor respiration may cause fatigue, fuzzy thinking, headaches, and susceptibility to pneumonia. (Respiratory insufficiency is a leading cause of death in ALS.)
- Speech, or eating and drinking by mouth, may not be possible
- Assistance needed if not needed in the previous stage yet; in the home care you would need:
- Power wheelchair, hospital bed, mechanical lift, and switches that enable any moving body part to operate computers, environmental control units, and communication devices.
- Assisted ventilation, either noninvasive or invasive (tracheostomy).
- Feeding tube.
- Possibly urinary catheters aren’t required but can make toileting easier.
- The type of home assistance you need:
1.) Caregivers should:
- Find and train caregiving help.
- Oversee 24-hour care operations.
- Find ways to help the person with ALS stay socially and mentally active.
- Get into a routine that supports themselves as well as the person with ALS.
- Know that although this stage is all-consuming, surprisingly many caregivers report great stability and satisfaction in their daily lives at this later stage of the disease.
2.) Visiting RN (Nurse) who basically follows up on the care and decline or no change in pt with letting the attending M.D. in charge be kept informed on pt’s status who makes any change with orders on the pt’s care. It’s a whole team effect to make sure the pt gets the best care!
- Progression is not always a straight line in an individual, either. It is not uncommon to have periods lasting weeks to months where there is very little or no loss of function. There are even very rare examples in which there is significant improvement and recovery of lost function. These ALS “arrests” and “reversals” are, unfortunately, usually transient. Less than 1 percent of people with ALS will have significant improvement in function lasting 12 months or longer
End stage
- The vast majority of deaths in ALS are the result of respiratory failure, a process that progresses slowly over months. Medications can relieve discomfort, anxiety, and fear caused by respiratory insufficiency.
- Far less-common causes of death in ALS include malnutrition as a result of swallowing problems, pulmonary embolism (a blockage in one of the arteries of the lungs), abnormalities in the heart’s electrical pacing system called cardiac arrhythmias, and pneumonia as the result of aspiration (when food or fluid gets into the lungs).
- Hospice care (in a facility or in the home) focuses on providing comfort and maintaining quality of life by supporting the physical, emotional, and spiritual needs of the individual with ALS and their family members. Families should contact hospice early on to see what in-home services are available even before the most advanced stage.
- At MDA clinics, physicians work closely with palliative care teams to coordinate treatment with in-home hospice care providers, assisted living facilities, or inpatient hospice settings. Such cooperation helps ensure the person with ALS has the most peaceful and painless experience possible.





 10Years Ago
10Years Ago