
What is Spinal Muscular Atrophy?
Spinal muscular atrophy is a genetic disorder characterized by weakness and wasting in muscles used for movement (skeletal muscles). It is caused by a loss of specialized nerve cells, called motor neurons that control muscle movement. In this disease we have an insufficient amount of SMN protein, which leads to permanent loss of motor neurons (Destruction). The weakness tends to be more severe in the muscles that are close to the center of the body (proximal) compared to muscles away from the body’s center (distal). The muscle weakness usually worsens with age. There are many types of spinal muscular atrophy that are caused by changes in the same genes. The types differ in age of onset and severity of muscle weakness; however, there is overlap between the types. Other forms of spinal muscular atrophy and related motor neuron diseases, such as spinal muscular atrophy with progressive myoclonic epilepsy, spinal muscular atrophy with lower extremity predominance, X-linked infantile spinal muscular atrophy, and spinal muscular atrophy with respiratory distress type 1 are caused by mutations in other genes.
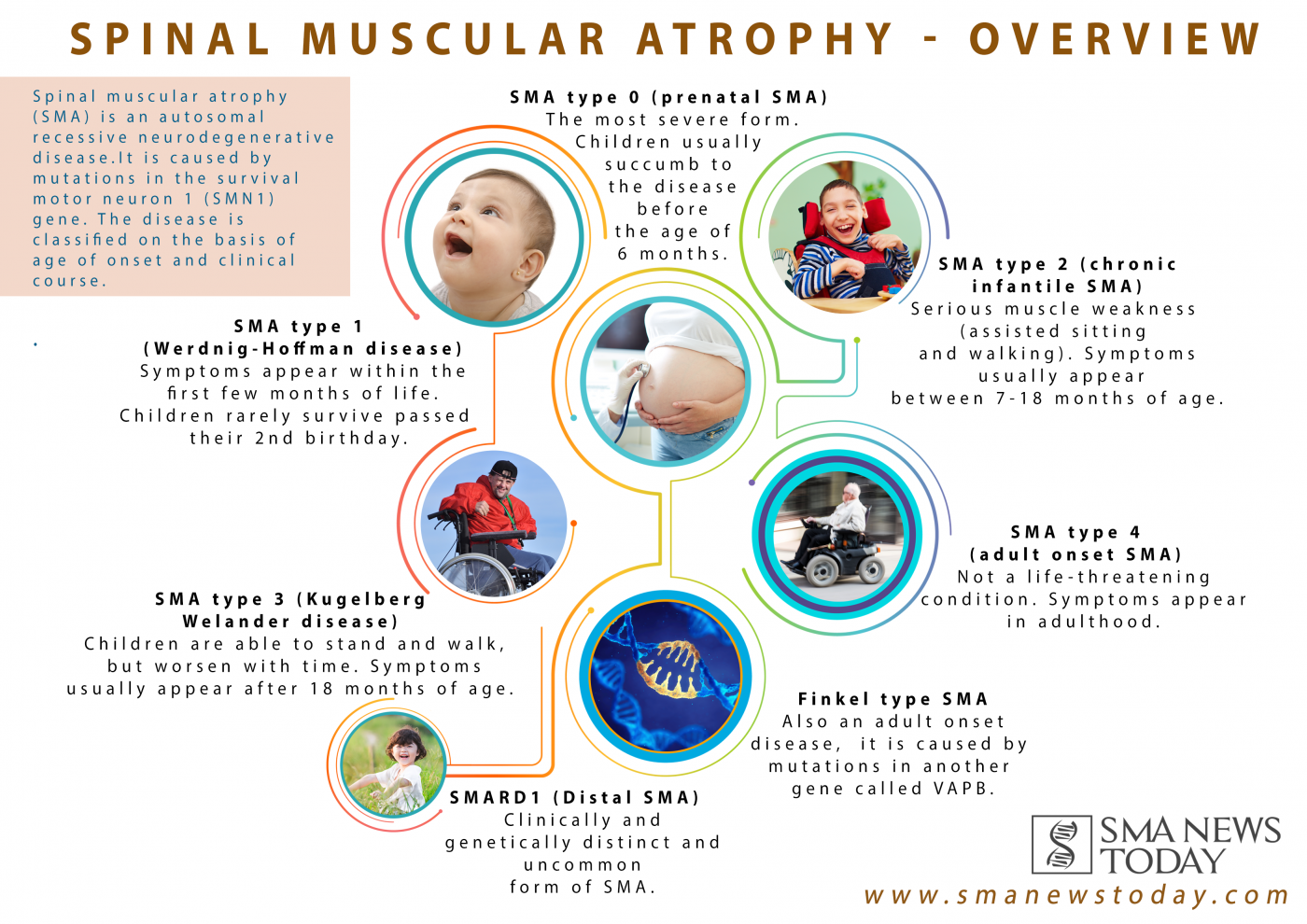
Spinal muscular atrophy type 0 is evident before birth and is the rarest and most severe form of the condition. Affected infants move less in the womb, and as a result they are often born with joint deformities (contractures). They have extremely weak muscle tone (hypotonia) at birth. Their respiratory muscles are very weak and they often do not survive past infancy due to respiratory failure. Some infants with spinal muscular atrophy type 0 also have heart defects that are present from birth (congenital).
STAGING OF SPINAL MUSCULAR ATROPHY:
Spinal muscular atrophy type I (also called Werdnig-Hoffmann disease) is the most common form of the condition. It is a severe form of the disorder with muscle weakness evident at birth or within the first few months of life. Most affected children cannot control their head movements or sit unassisted. Children with this type may have swallowing problems that can lead to difficulty feeding and poor growth. They can also have breathing problems due to weakness of respiratory muscles and an abnormally bell-shaped chest that prevents the lungs from fully expanding. Most children with spinal muscular atrophy type I do not survive past early childhood due to respiratory failure.
Spinal muscular atrophy type II (also called Dubowitz disease) is characterized by muscle weakness that develops in children between ages 6 and 12 months. Children with this type can sit without support, although they may need help getting to a seated position. However, as the muscle weakness worsens later in childhood, affected individuals may need support to sit. Individuals with spinal muscular atrophy type II cannot stand or walk unaided. They often have involuntary trembling (tremors) in their fingers, a spine that curves side-to-side , and respiratory muscle weakness that can be life-threatening. The life span of individuals with spinal muscular atrophy type II varies, but many people with this condition live into their twenties or thirties.
Spinal muscular atrophy type III (also called Kugelberg-Welander disease) typically causes muscle weakness after early childhood. Individuals with this condition can stand and walk unaided, but over time, walking and climbing stairs may become increasingly difficult. Many affected individuals require wheelchair assistance later in life. People with spinal muscular atrophy type III typically have a normal life expectancy.
Spinal muscular atrophy type IV is rare and often begins in early adulthood. Affected individuals usually experience mild to moderate muscle weakness, tremors, and mild breathing problems. People with spinal muscular atrophy type IV have a normal life expectancy.
JOHN HOPKINS MEDICINE states these types of SMA:
| Type | Age at onset | Symptoms, rate of progression, and life expectancy |
|---|---|---|
| Becker | adolescence to early adulthood | Symptoms are almost identical to Duchenne, but less severe; progresses more slowly than Duchenne; survival into middle age. As with Duchenne, disease is almost always limited to males. |
| Congenital | birth | Symptoms include general muscle weakness and possible joint deformities; disease progresses slowly; shortened life span. |
| Duchenne | 2 to 6 years | Symptoms include general muscle weakness and wasting; affects pelvis, upper arms, and upper legs; eventually involves all voluntary muscles; survival beyond 20s is rare. Seen in boys only. Very rarely can affect woman, who have much milder symptoms and a better prognosis. |
| Distal | 40 to 60 years | Symptoms include weakness and wasting of muscles of the hands, forearms, and lower legs; progression is slow; rarely leads to total incapacity. |
| Emery-Dreifuss | childhood to early teens | Symptoms include weakness and wasting of shoulder, upper arm, and shin muscles; joint deformities are common; progression is slow; sudden death may occur from cardiac problems. |
| Facioscapulohumeral | childhood to early adults | Symptoms include facial muscle weakness and weakness with some wasting of shoulders and upper arms; progression is slow with periods of rapid deterioration; life span may be many decades after onset. |
| Limb-Girdle | late childhood to middle age | Symptoms include weakness and wasting, affecting shoulder girdle and pelvic girdle first; progression is slow; death is usually due to cardiopulmonary complications. |
| Myotonic | 20 to 40 years | Symptoms include weakness of all muscle groups accompanied by delayed relaxation of muscles after contraction; affects face, feet, hands, and neck first; progression is slow, sometimes spanning 50 to 60 years. |
| Oculopharyngeal | 40 to 70 years | Symptoms affect muscles of eyelids and throat causing weakening of throat muscles, which, in time, causes inability to swallow and emaciation from lack of food; progression is slow. |