![]()
SCD is the most commonly inherited blood disorder in the United States, affecting 100,000 people, and millions more worldwide. The disease primarily affects people of African, Hispanic, Mediterranean, Middle Eastern and South Asian ancestry.
What is sickle cell disease actually?
The term sickle cell disease (SCD) describes a group of inherited red blood cell disorders. People with SCD have abnormal hemoglobin, called hemoglobin S or sickle hemoglobin, in their red blood cells.
Hemoglobin is a protein in red blood cells that carries oxygen throughout the body.
“Inherited” means that the disease is passed by genes from parents to their children. SCD is not contagious. A person cannot catch it, like a cold or infection, from someone else.
People who have SCD inherit two abnormal hemoglobin genes, one from each parent. In all forms of SCD, at least one of the two abnormal genes causes a person’s body to make hemoglobin S. When a person has two hemoglobin S genes, Hemoglobin SS, the disease is called sickle cell anemia. This is the most common and often most severe kind of SCD.
Hemoglobin SC disease and hemoglobin Sβ thalassemia (thal-uh-SEE-me-uh) are two other common forms of SCD.
Cells in tissues need a steady supply of oxygen to work well. Normally, hemoglobin in red blood cells takes up oxygen in the lungs and carries it to all the tissues of the body.
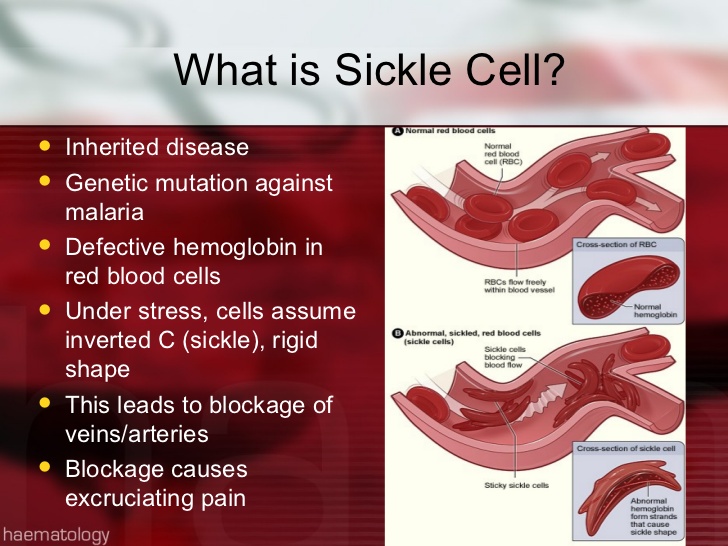
Red blood cells that contain normal hemoglobin are disc shaped (like a doughnut without a hole). This shape allows the cells to be flexible so that they can move through large and small blood vessels to deliver oxygen.
Sickle hemoglobin is not like normal hemoglobin. It can form stiff rods within the red cell, changing it into a crescent, or sickle shape.
Sickle-shaped cells are not flexible and can stick to vessel walls, causing a blockage that slows or stops the flow of blood. When this happens, oxygen can’t reach nearby tissues.
The lack of tissue oxygen can cause attacks of sudden, severe pain, called pain crisis. These pain attacks can occur without warning, and a person often needs to go to the hospital for effective treatment.
Most children with SCD are pain free between painful crises, but adolescents and adults may also suffer with chronic ongoing pain.
The red cell sickling and poor oxygen delivery can also cause organ damage. Over a lifetime, SCD can harm a person’s spleen, brain, eyes, lungs, liver, heart, kidneys, penis, joints, bones, or skin.
Sickle cells can’t change shape easily, so they tend to burst apart or hemolyze. Normal red blood cells live about 90 to 120 days, but sickle cells last only 10 to 20 days.
The body is always making new red blood cells to replace the old cells; however, in SCD the body may have trouble keeping up with how fast the cells are being destroyed. Because of this, the number of red blood cells is usually lower than normal. This condition, called anemia, can make a person have less energy. Anemia ending line is lack of oxygen to the tissue body parts all over.
“Sickle cell disease is devastating for patients and their families,” said Jeffrey Glassberg, MD, MA, Assistant Professor of Emergency Medicine, Hematology and Medical Oncology, Icahn School of Medicine at Mount Sinai. “It’s a chronic disorder causing pain in the extremities and back, infections, organ failure and other tissue damage, skin infections, loss of eyesight, severe blood clots and strokes. Patients learn to function in a constant state of pain and when that pain becomes debilitating, they often end up in the emergency room,” said Dr. Glassberg, also Associate Director of the Comprehensive Sickle Cell Program at The Mount Sinai Hospital.
“Patients with SCD are more likely to live full lives if they undergo regular checkups, prevent infections and develop healthy habits,” said Jena Simon, MS, FNP-BC, RN, also of the Comprehensive Sickle Cell Program.
Tips to Staying Healthy
- Get regular checkups. Regular health checkups can help prevent some serious problems.
- Prevent infections. Common illnesses, like influenza quickly can become dangerous for both children and adults with SCD. The best defense is to get a flu shot every fall and to stay up-to-date on other immunizations.
- People with SCD should drink 8 to 10 glasses of water every day and eat healthy food. They also should try not to get too hot, too cold, or too tired.
- Look for clinical studies. New clinical research studies are beginning all the time at Mount Sinai and elsewhere, with the goal of finding better treatments for SCD. Study participants gain early access to experimental medicines and treatments.
- Get support. People with SCD should find a patient support group or other organization in the community that can provide information, assistance, and support.
Sickle Cell Disease Facts & Figures:
- SCD is an inherited blood disorder that can cause severe pain and permanent damage to the brain, heart, lungs, kidneys, liver, bones and spleen.
- SCD is most common in Africans and African-Americans. It is also found in other ethnic and racial groups, including people from South and Central America, the Caribbean, Mediterranean countries, and India.
- More than 2 million people carry the sickle cell gene that allows them potentially to pass the disease on to their children. People of African, Hispanic, Mediterranean, Middle Eastern, and Indian descent may want to be tested for the gene before having children. You can carry the gene and not have any signs or symptoms of SCD. Both parents have to have the gene to have a child with SCD.