For animals that hibernate, making it to spring is no small feat. Torpor — the state of reduced bodily activity that occurs during hibernation — is not restful. By the time they emerge, hibernating animals are often sleep-deprived: Most expend huge bursts of energy to arouse themselves occasionally in the winter so their body temperatures don’t dip too low. This back-and-forth is exhausting, and hibernators do it with little to no food and water. By winter’s end, some have shed more than half their body weight.
But just because it’s spring doesn’t mean it’s time to celebrate. Spring means getting ready for the full speed of summer — and
With the onset of spring in the Northern Hemisphere, animals that hibernate are waking up from a long-period of deep sleep. They spent the winter hibernating to conserve energy when food was scarce. Animals that hibernate include bats, black bears, Arctic ground squirrels, and common poorwill birds. Many other species such as raccoons and skunks go into a state of torpor during the cold weather, which is a type of light hibernation. Most hibernators wake up during the months of March and April, but some do so as late as May.
This would include the following creatures:
1.) Bats 

Many types of bats hibernate through the long, cold winter in caves. Bats that hibernate include the little brown bat, the big brown bat, and the northern long-eared bat. During hibernation, their body temperature, heart rate, breathing rate, and metabolism drop to very low levels. This allows them to get by without food or water and stay in a dormant state for long periods of time.
Fertilization happens a few days after females emerge from hibernation. After leaving their winter caves, they move to a large tree or another cave. “They want a warm, stable environment where they can develop their young,” said Joy M. O’Keefe, a bat expert and assistant professor at Indiana State University.
Bats often return to the same maternity spot year after year, sometimes traveling hundreds of miles to get there. Dozens of mothers will congregate at these sites, cuddling to keep warm. When their pups are born, 50 to 60 days later, mothers may help each other by taking turns foraging for insects and roosting with the group. With no parenting responsibilities, and perhaps to avoid competing with the females, males will stay in torpor for longer — making their hibernation spaces real man caves in the spring.
As spring arrives, so do bats! Many naturalists state during this season looking for migrating salamanders and blossoming bloodroot. They never thought much about what bats are doing this time of year.
It turns out these flying mammals, who retreated into hibernation back in the fall, are emerging from April through May, as the weather grows consistently warmer and insects again fill the air.
2.) Bears 

When spring arrives and the snow begins to melt, bears start to wake up after months of hibernation. It is an exciting time of the year for bears and park visitors. When bears emerge from their dens, understandably hungry, they immediately begin to search for food. And there is plenty to eat. Receding snow reveals vegetation rich in nutrients. Winter kill – deer, elk, moose or anything else that may fancy a bear’s taste buds, are easy pickings. It’s an important time of the year for a bear as it begins the process of nourishing itself, continually gorging on food throughout the year in preparation for hibernation in the fall. For visitors beginning their spring and summer vacations, the emergence of bears means a chance to see a bruin in its natural habitat, its home. But it also means that another food source presents itself to bears – the food you may accidentally (or intentionally) leave behind or provide. Storing your food and disposing of garbage properly can mean life or death to a bear. Be sure to always properly store food in bear country.
One of the many reasons people visit national parks with bears is to experience a wild place capable of supporting healthy populations of black and grizzly bears. When visitors become careless and do not properly store their food, bears are undoubtedly going to find it; their sense of smell is amazing. When visitors feed bears, it’s a recipe for trouble. If bears become used to approaching people and eating human food (we call that habituation), the bear no longer seeks the natural food it is supposed to be foraging for. This creates a management and safety problem for park visitors and bears. While park staff work to manage bears and visitors, sometimes there is a need to remove a bear from a park. Imagine what that does to the ecosystem and your experience as a visitor coming to see a bear. For many, it means the park experience is diminished, and the ecosystem isn’t as intact.
When we visit a park with bears, we are entering their home. As guests, proper behavior and etiquette on our part can contribute to a safe and enjoyable visit for us as our hosts.
3.) Arctic Squirrels: 

Arctic ground squirrels are the largest of the North American ground squirrel species, ranging from 524 up to 1,500 grams in weight, and 332 to 495 mm in length. They undergo seasonal changes in body mass and lose weight during hibernation. They exhibit sexual dimorphism, with males being larger than females. Body mass drastically varies seasonally, between summer foraging bouts and winter hibernation. They have tawny brown coloration with white flecks on the dorsal side of the pelage and a light tan or beige coloration on their undersides. Their undersides lighten during winter months.
During the onset of cold weather, Arctic ground squirrels dig deep burrows in the ground and hibernate. One scientist attached temperatures sensors to their abdomens and recorded body temperatures in hibernating squirrels as low as -2.9 degrees Celsius (26.8 degrees Fahrenheit), which is below the temperature that water freezes! The squirrel’s blood, however, does not freeze in part because it is salty and also because they have some sort of “super cool” supercooling mechanism that protects them. Scientists are actively researching the brain activity of hibernating Arctic ground squirrels for insights into how to protect people from neurodegenerative diseases like Alzheimer’s and to help them recover from brain injuries. Specifically, the brains of Arctic ground squirrels show a remarkable ability to bounce back after months of dormancy that degrades neuronal connections.
Arctic ground squirrels generally begin hibernation in the beginning of August and wake up in early April, when the males dig their way out from underground.
4.) Common poorwill 

Most birds migrate south when the weather turns cold, but the common poorwill stays put and hibernates. Poorwills are the only bird species known to hibernate. They can be found in the western United States and Canada. Native Americans often referred to this bird as “the sleeping one.”
5.) Torpor = Racco0n and Skunk
Torpor is a state of light hibernation that many animals enter into to survive the winter. Animals that use torpor as a survival strategy include raccoons and skunks. 
While there is no bright line that separates animals that hibernate from those that use torpor, it generally comes down to the length of time that an animal spends in dormancy and the extent to which its body temperature and metabolic rate are depressed. Torpor is associated with brief periods of dormancy, sometimes for only a few hours, and small physiological changes, whereas hibernation is associated with lengthy periods of dormancy and large physiological changes.
6.) Reptiles:
Scientists use the term brumation to refer to hibernating-like states in reptiles,

which are not warm-blooded animals so the physiological responses are a bit different from those in mammals and birds. Insects enter cold-induced dormant periods too, and this is referred to by the term diapause. Often on the internet, the term hibernation will be used as a catch-all phrase for all of these types of dormant states.
The exact triggers that cause an animal to enter into and emerge from hibernation aren’t well known, but combinations of factors such as changes in temperature, daylight, and food availability are thought to play an important role. Especially critical is an animal’s internal biological clock, which will initiate hormone changes when it is time for the animal to wake up.
Bottom line: Hibernation is a survival strategy that animals use during the winter to conserve energy when food is scarce. Animals that hibernate include bats, black bears, Arctic ground squirrels, and common poorwill birds.











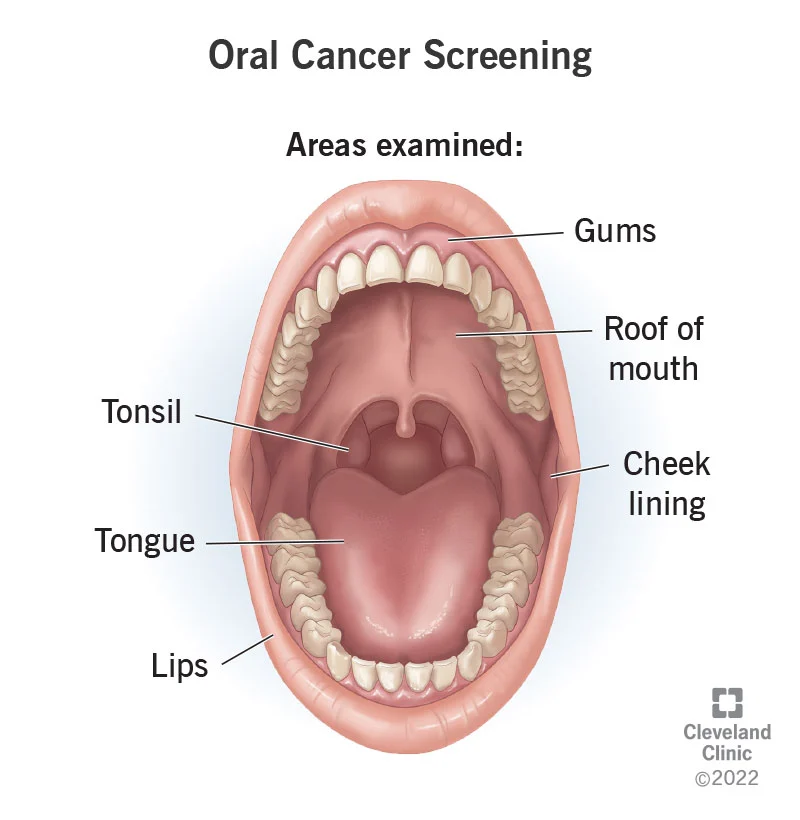
 ORAL CANCER RECONCONSTRUCTION SURVIVORS
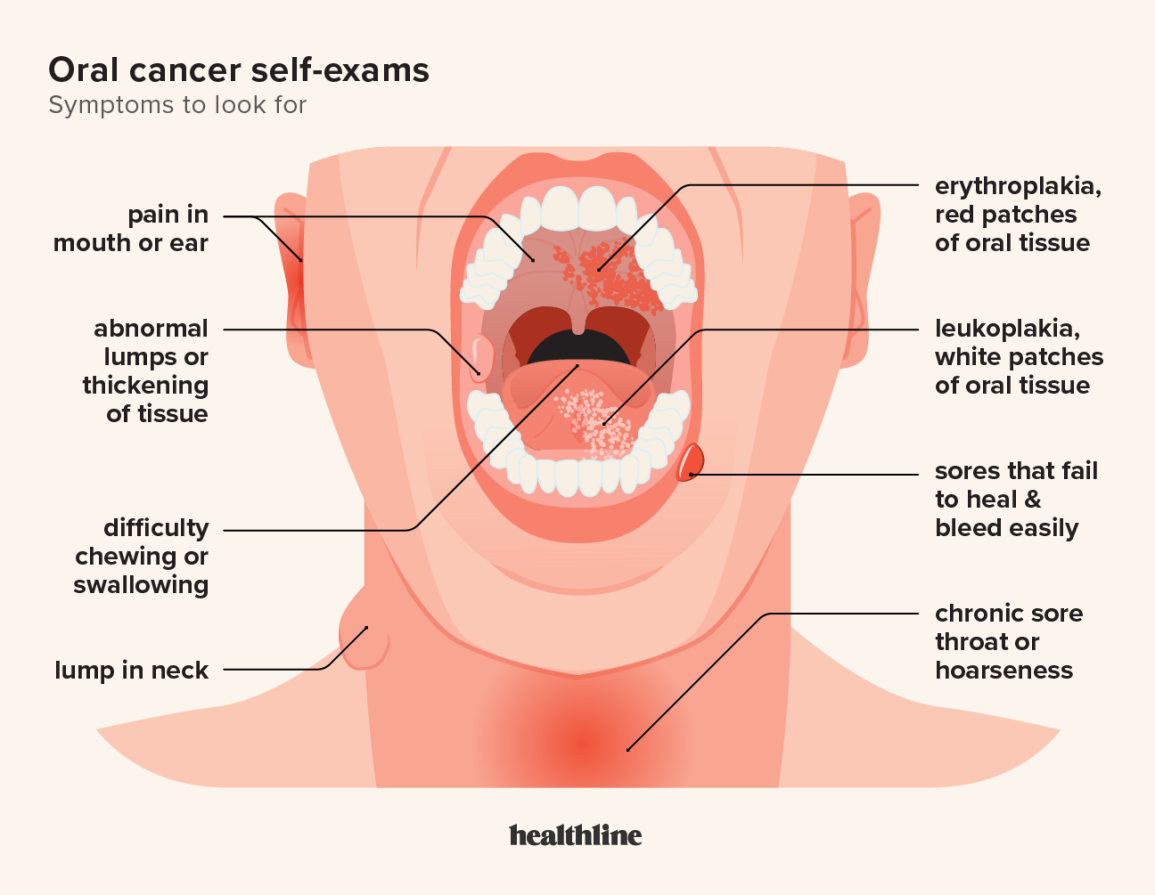
ORAL CANCER RECONCONSTRUCTION SURVIVORS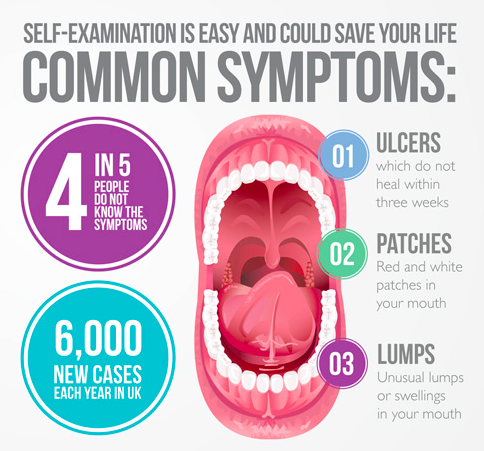 KNOW THE SYMPTOMS
KNOW THE SYMPTOMS